
2024年4月18日の令和6年度第1回厚生科学審議会医薬品医療機器制度部会から、次期薬機法改正の議論が開始されています(関連記事)。
2024年5月17日の令和6年度第2回会議では業界要望の確認がされました(関連記事)。
2024年6月6日に、令和6年度第3回会議が開催となりました。この会議では、テーマごとの検討として、「①ドラッグロスや供給不足などの医薬品等へのアクセスの課題に対応した安全かつ迅速な承認制度の確立」が議論されていましたので、主なポイントを整理しておきます。
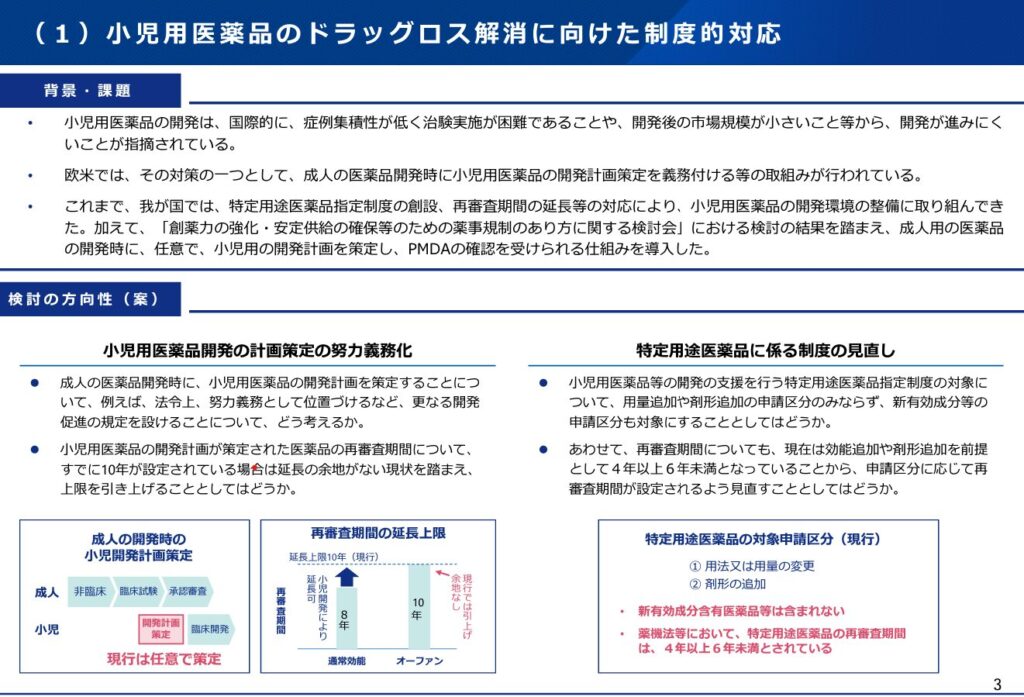
小児用医薬品の開発
「創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会」の結果を受けて、小児用医薬品の開発推進については、PMDAによる開発計画の確認の基づく薬価上の手当等一定の対応がすでに実施されています。
今回の薬機法改正の議論での追加事項としては、小児開発の努力義務化、再審査期間の10年を超える延長が大きなポイントとなりそうです。欧米でも義務とされつつも免除の品目や開発がなかなか進まない品目があることから、努力義務がどの程度の効果を示すかは疑問があります。また、再審査期間の10年を超える延長もどこまで効果があるかは疑問がありますが(日本の先発品独占期間は再審査期間と特許期間のいずれか長いほうになるため)、こちらのほうが開発推進の寄与の可能性は高いのではないかと考えます。
中等度変更事項の導入
製造管理関連ですが、「創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会」の結果に沿った形で法的手当が提示されました。ここについては、大きな議論なく、導入に向けた法改正作業が進みそうです。

GMP適合性調査の見直し
新薬でのGMP適合性調査は原則実地、定期GMP調査での製造業者による申告(に基づくリスクベースの調査)という合理化(メリハリ対応)の方向性が示されました。
一方で、GMPの適合性が品目の承認に紐づくという建付け自体は維持されそうです。

再生医療等製品の規格外品
自家細胞品(CART製品等)の再生医療等製品について、含量等の規格外品を製造販売可能とする仕組みが新たに導入されそうです。こちらは、前回会議の業界要望のうち、再生医療イノベーションフォーラム(FIRM)が提案していた内容が概ね反映されています。

その他
条件付き承認の可能性の模索も論点となっていますが、具体的な手当はさほどなさそうであり、また、薬機法自体の改正が不可欠な内容までには至っていないようです。
リアルワールドデータ(RWD)の承認申請での活用は、法的位置づけとしてそのようなデータが利用可能であることが明示的となるよう、条文の手当が挙げられていますが、実態(運用)上は影響がないと考えます。結局、どのようなデータがあり、承認審査に使えるかは引き続き個別に判断となる可能性が高いです。
関連リンク
- 医薬品医療機器制度部会のページ:https://www.mhlw.go.jp/stf/shingi/shingi-kousei_430263.html
- 令和6年度第3回医薬品医療機器制度部会のページ(各種資料あり):https://www.mhlw.go.jp/stf/newpage_40580.html
- 創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会:https://www.mhlw.go.jp/stf/shingi/other-iyaku_128701_00006.html
- 2025年薬機法改正に向けた議論の開始:https://regulatory-j.com/act-revision-2024-1/
- 業界要望:2025年薬機法改正に向けた議論:https://regulatory-j.com/act-revision-2/
- 医薬品の製造方法等に係る薬事審査等のあり方:https://regulatory-j.com/manufacturing-change/

コメント