
ドラッグ・ラグ/ドラッグ・ロスの解消や創薬力強化を目指して薬事規制関連の議論を進めるために、「創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会」が2023年5月~2024年3月に開催されてきました。
この検討会で議論があり、引き続き、検討となっている事項に製造方法(の変更)に関する承認の話があります。
以下、検討会の報告書にある図になりますが、製造方法の変更に対する日米欧の規制対応がきれいに整理されています。
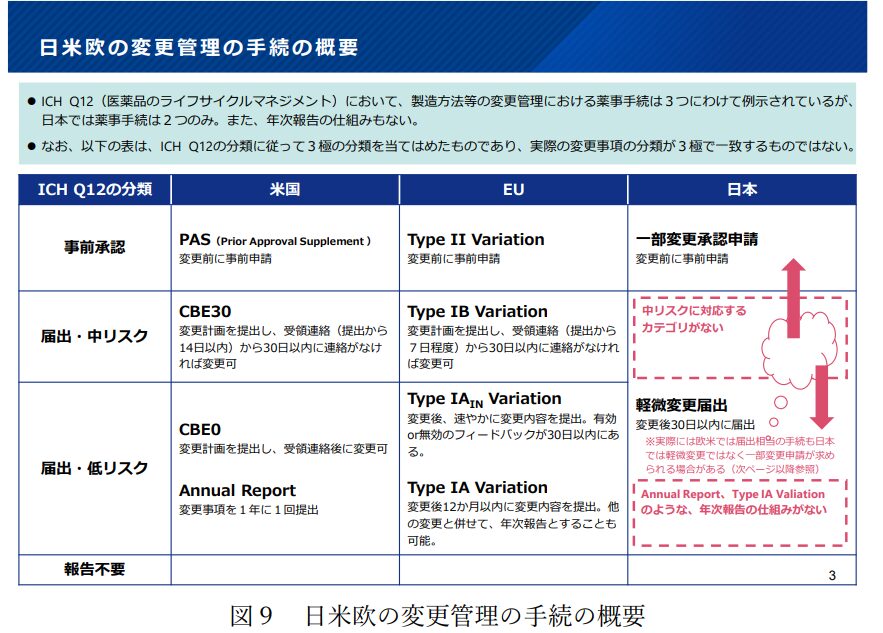
日本では製造方法の変更にあたって、その変更内容の程度に応じて、承認事項の一部変更承認申請(一変申請)か軽微変更届出の規制対応が必要になります。
一変申請の場合は、実際に製造方法の変更を導入する前に、PMDAの審査、MHLWの承認を受ける必要があります。したがって、製造方法の変更を行うまでに相応の期間(通常の標準審査期間は12か月)を要することになります。
軽微変更届出の場合は、届出であることからPMDAの審査やMHLWの承認といった対応は発生しません。また、先に製造方法の変更を導入し、届出は変更から30日以内の提出となります。ただし、実際には、製造方法の変更内容が軽微変更届出に該当するかどうかを、別途PMDAに相談して対応する必要が多々あります。
このような現状を踏まえて、検討会で結論を得た方向性として、中等度変更事項の導入、年次報告の導入という新たな仕組みが提示されています。
中等度変更事項の導入
こちらはステップを踏んでの導入が想定されています。対象を限定しての試行を行って様子をみようとしています。
変更内容のリスクに応じて、変更内容を事前かつ短期間で確認することを想定した仕組みになります。そして、初回承認申請又は一変申請の審査においてあらかじめ「中等度変更事項」として特定する場合と、変更が生じた場合に都度PMDA相談で中等度変更事項への該当性を確認していく場合が想定されています。
この変更手続きは、一変申請の枠組みの中で対応することとされています。したがって、中等度変更事項の導入では、引き続き(期間は短くなるものの)一変申請が必要となってきます。また、PMDA相談も引き続き必要となる事例が残っていくことになります。
そのため、中等度変更事項では、先の申請の中で妥当性が確認されていれば予見可能性は高まるものの、製造方法の変更にあたって、事前に一変申請が必要ということで、今の薬事制度の延長のようなイメージです。
年次報告の導入
中等度変更事項とは異なり、こちらは新たな仕組みとなります。重要度の低い変更内容(現状の軽微変更届の対象となっている事項を含む。)について、希望する場合に、年次報告で対応できるようにしようとするものです。
年次報告の内容確認にあたっては、 PMDA の相談の枠組みを活用する可能性が検討会の報告書中に記載されています。新たな仕組みであり、確認にあたってPMDAのリソースをどう確保するかに対して手を打っていると考えます。希望する場合かつ相談の枠組みとすることで、手数料収入によるPMDAのリソース確保を想定していそうです。
中等度変更事項、年次報告の導入方法、時期
検討会の報告書では、行政・業界間で具体的な検討を進めていくとされています。しっかりした制度の導入にあたっては、これらの事項は薬機法の改正を伴う可能性が高そうです。
実際、2025年薬機法改正に向けた議論で触れましたが、薬機法改正の議論テーマ・論点の中に「製造方法等の中リスクの変更カテゴリの追加」が挙げられています。
薬機法改正の議論は、2024年7月に目途で整理され、年内を目途に取りまとめることとされています。したがって、早ければ2025年1月からの通常国会に薬機法改正に関する法案が提出される可能性があります。

コメント